


 Sign Out
Sign Out
The frequencies of adverse events are ranked according to the following: Very common (≥ 1/10), common (≥ 1/100, < 1/10), uncommon (≥1/1000, < 1/100), rare (≥ 1/10,000, < 1/1000), very rare (<1/10,000) including isolated reports, not known (cannot be estimated from the available data).
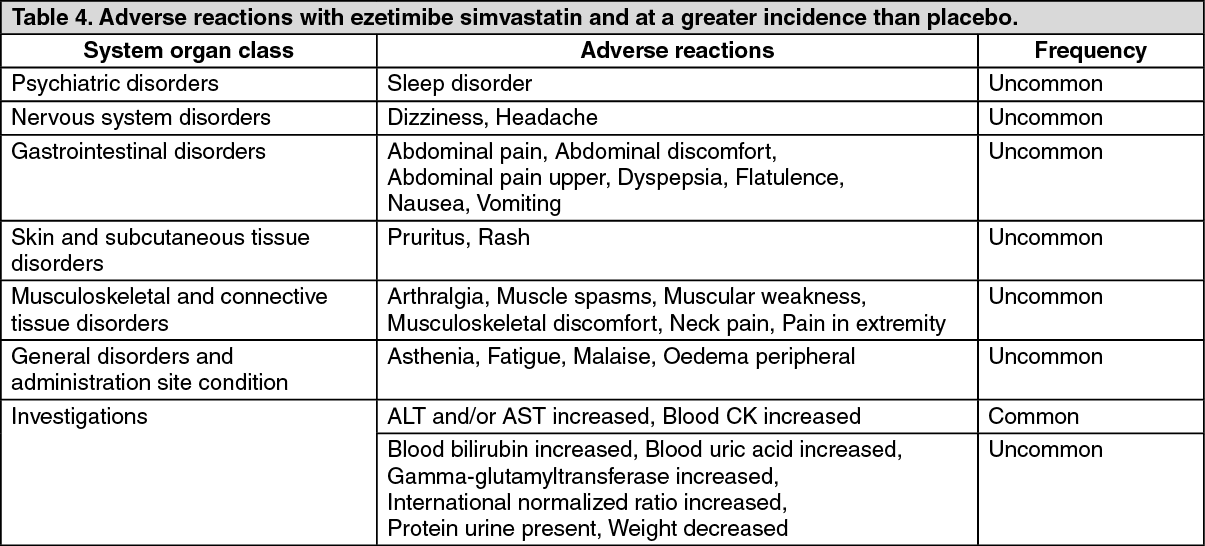
The following adverse reactions were observed in patients treated with ezetimibe simvastatin (N=2404) and at a greater incidence than placebo (N=1340). (See Table 4.)
 Click on icon to see table/diagram/image
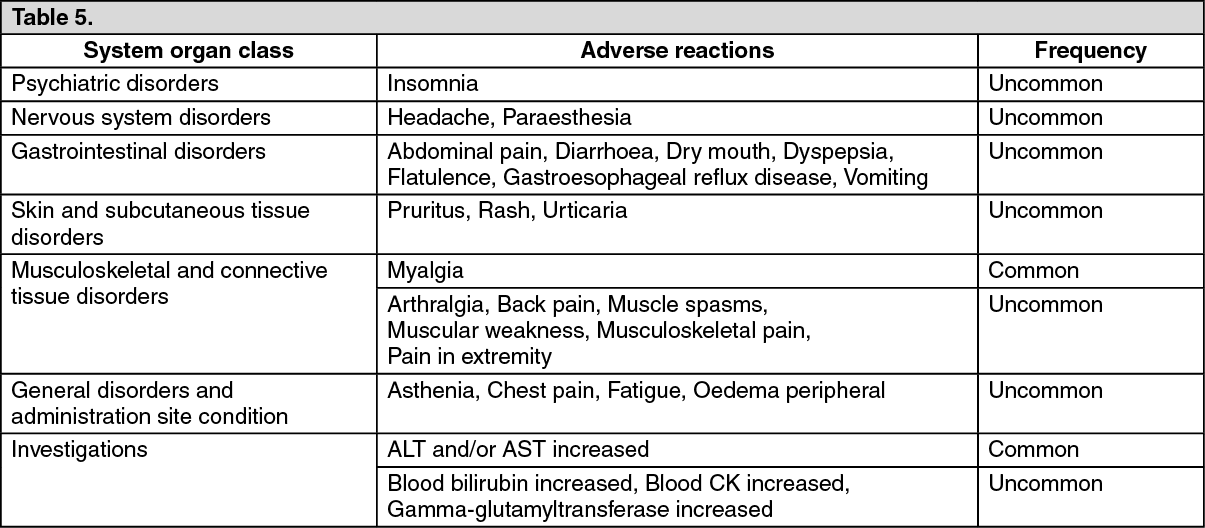
Click on icon to see table/diagram/imageThe following adverse reactions were observed in patients treated with Co-Ezetimibe TEVA (N=9595) and at a greater incidence than statins administered alone (N=8883). (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePaediatric population: In a study involving adolescent (10 to 17 years of age) patients with heterozygous familial hypercholesterolaemia (n = 248), elevations of ALT and/or AST (≥3X ULN, consecutive) were observed in 3% (4 patients) of the ezetimibe simvastatin patients compared to 2% (2 patients) in the simvastatin monotherapy group; these figures were respectively 2% (2 patients) and 0% for elevation of CPK (≥ 10X ULN). No cases of myopathy were reported.
This trial was not suited for comparison of rare adverse drug reactions.
Patients with Coronary Heart Disease and ACS Event History: In the IMPROVE-IT study (see PHARMACOLOGY: Pharmacodynamics under Actions), involving 18,144 patients treated with either Co-Ezetimibe TEVA 10/40 mg (n=9067; of whom 6% were uptitrated to Co-Ezetimibe TEVA 10/80 mg) or simvastatin 40 mg (n=9077; of whom 27% were uptitrated to simvastatin 80 mg), the safety profiles were similar during a median follow-up period of 6.0 years. Discontinuation rates due to adverse experiences were 10.6% for patients treated with ezetimibe simvastatin and 10.1% for patients treated with simvastatin. The incidence of myopathy was 0.2% for Co-Ezetimibe TEVA and 0.1% for simvastatin, where myopathy was defined as unexplained muscle weakness or pain with a serum CK ≥ 10 times ULN or two consecutive observations of CK ≥5 and <10 times ULN. The incidence of rhabdomyolysis was 0.1% for ezetimibe simvastatin and 0.2% for simvastatin, where rhabdomyolysis was defined as unexplained muscle weakness or pain with a serum CK ≥10 times ULN with evidence of renal injury, ≥5 times ULN and <10 times ULN on two consecutive occasions with evidence of renal injury or CK ≥10,000 IU/L without evidence of renal injury. The incidence of consecutive elevations of transaminases (≥3 X ULN) was 2.5% for ezetimibe simvastatin and 2.3% for simvastatin. (See Precautions.) Gallbladder-related adverse effects were reported in 3.1% vs 3.5% of patients allocated to Co-Ezetimibe TEVA and simvastatin, respectively. The incidence of cholecystectomy hospitalisations was 1.5% in both treatment groups. Cancer (defined as any new malignancy) was diagnosed during the trial in 9.4% vs 9.5%, respectively.
Patients with Chronic Kidney Disease: In the Study of Heart and Renal Protection (SHARP) (see PHARMACOLOGY: Pharmacodynamics under Actions), involving over 9000 patients treated with Co-Ezetimibe TEVA 10 mg/20 mg daily (n=4650) or placebo (n=4620), the safety profiles were comparable during a median follow-up period of 4.9 years. In this trial, only serious adverse events and discontinuations due to any adverse events were recorded. Discontinuation rates due to adverse events were comparable (10.4% in patients treated with Co-Ezetimibe TEVA, 9.8% in patients treated with placebo). The incidence of myopathy/rhabdomyolysis was 0.2% in patients treated with Co-Ezetimibe TEVA and 0.1% in patients treated with placebo. Consecutive elevations of transaminases (> 3X ULN) occurred in 0.7% of patients treated with Co-Ezetimibe TEVA compared with 0.6% of patients treated with placebo. In this trial, there were no statistically significant increases in the incidence of pre-specified adverse events, including cancer (9.4% for ezetimibe simvastatin, 9.5% for placebo), hepatitis, cholecystectomy or complications of gallstones or pancreatitis.
Laboratory Values: In coadministration trials, the incidence of clinically important elevations in serum transaminases (ALT and/or AST ≥ 3 X ULN, consecutive) was 1.7% for patients treated with Co-Ezetimibe TEVA.
These elevations were generally asymptomatic, not associated with cholestasis, and returned to baseline after discontinuation of therapy or with continued treatment. (See Precautions.)
Clinically important elevations of CK (≥ 10 X ULN) were seen in 0.2% of the patients treated with ezetimibe simvastatin.
Post-marketing Experience: The following additional adverse reactions have been reported in post-marketing use with ezetimibe simvastatin or during clinical studies or post-marketing use with one of the individual components.
Blood and lymphatic system disorders: thrombocytopaenia; anaemia.
Nervous system disorders: peripheral neuropathy; memory impairment.
Respiratory, thoracic and mediastinal disorders: cough; dyspneoa, interstitial lung disease (see Precautions).
Gastro-intestinal disorders: constipation; pancreatitis; gastritis.
Skin and subcutaneous tissue disorders: alopecia; erythema multiforme; rash; urticaria; angioedema.
Immune system disorders: hypersensitivity, including anaphylactic reactions; anaphylaxis (very rare).
Musculoskeletal and connective tissue disorders: muscle cramps; myopathy* (including myositis); rhabdomyolysis with or without acute renal failure (see Precautions); tendinopathy, sometimes complicated by rupture; immune-mediated necrotizing myopathy (IMNM) (frequency not known)**.
* In a clinical trial, myopathy occurred commonly in patients treated with simvastatin 80 mg/day compared to patients treated with 20 mg/day (1.0% vs 0.02%, respectively) (see Precautions and Interactions).
**There have been very rare reports of immune-mediated necrotizing myopathy (IMNM), an autoimmune myopathy, during or after treatment with some statin. IMNM is clinically characterized by: persistent proximal muscle weakness and elevated serum creatine kinase, which persist despite discontinuation of statin treatment; muscle biopsy showing necrotizing myopathy without significant inflammation; improvement with immunosuppressive agents (see Precautions).
Metabolism and nutrition disorders: decreased appetite.
Vascular disorders: hot flush; hypertension.
General disorders and administration site conditions: pain.
Hepato-biliary disorders: hepatitis/jaundice; fatal and non-fatal hepatic failure; cholelithiasis; cholecystitis.
Reproductive system and breast disorders: erectile dysfunction.
Psychiatric disorders: depression; insomnia.
An apparent hypersensitivity syndrome has been reported rarely which has included some of the following features: angio-oedema, lupus-like syndrome, polymyalgia rheumatica, dermatomyositis, vasculitis, thrombocytopaenia, eosinophilia, red blood cell sedimentation rate increased, arthritis and arthralgia, urticaria, photosensitivity reaction, pyrexia, flushing, dyspnoea and malaise.
Laboratory Values: elevated alkaline phosphatase; liver function test abnormal.
Increases in HbA1c and fasting serum glucose levels have been reported with statins, including simvastatin.
There have been rare post-marketing reports of cognitive impairment (e.g. memory loss, forgetfulness, amnesia, memory impairment, confusion) associated with statin use, including simvastatin. The reports are generally nonserious, and reversible upon statin discontinuation, with variable times to symptom onset (1 day to years) and symptom resolution (median of 3 weeks).
The following additional adverse events have been reported with some statins: sleep disturbances, including nightmares; sexual dysfunction; Diabetes mellitus: Frequency will depend on the presence or absence of risk factors (fasting blood glucose ≥ 5.6 mmol/L, BMI > 30 kg/m2, raised triglycerides, history of hypertension).
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
View ADR Monitoring Form